
Regulatory Landscape - Overview
Neurological device Regulatory Landscape: Product Overview
Neurological devices are medical devices intended to diagnose, monitor or for the treatment of the various health conditions related to the nervous system of the body, which include brain, spinal cord, and peripheral nerves.
According to the Food and Drug Administration (FDA) panel for Neurological devices, “Neurological Devices” are defined as devices which support, monitor, or replace neurological functions in patients affected by neurological disorders or injuries.
Neurological device Types
Neurological devices used for the treatment of neurological health issues include cerebrospinal fluid (CSF) management devices, interventional neurology devices, neurosurgical devices and neurostimulation devices.
Neurological devices Applications
This device can be used for diagnostic purposes such as imaging or measuring electrical activity. therapeutic intervention such as neurostimulators or implantable devices for epilepsy or Parkinson’s disease. Rehabilitation includes periprosthetic and functional electrical stimulation (FES) systems. Surgical tools specific to neurological procedures.
Interventional neurology devices are used to diagnose and treat vascular diseases of the brain and central nervous system. Interventional neurology includes endovascular, catheter-based techniques, angiography, and fluoroscopy. Catheter angiography is one of the oldest primary in-vivo brain vascular imaging modalities used to diagnose several neurological conditions such as cerebral aneurysm, arteriovenous malformations, intracranial stenosis, arteriovenous fistula, and vasculitis.
Additionally, minimally invasive medical procedures provide significant clinical advantages over conventional surgical procedures, including shorter hospital stays, higher procedural safety & efficacy, faster patient recovery, and greater affordability. Thus, the market approval and demand for neurology devices and different minimally invasive surgical procedures is increasing among medical professionals across the world.
Neurological device Product Development steps:
Centre for devices and radiological health (CDRH) is responsible for ensuring the safety, efficacy and quality of the medical devices including neurological devices intended for the treatment of neurological disorders under food and drug administration (FDA).
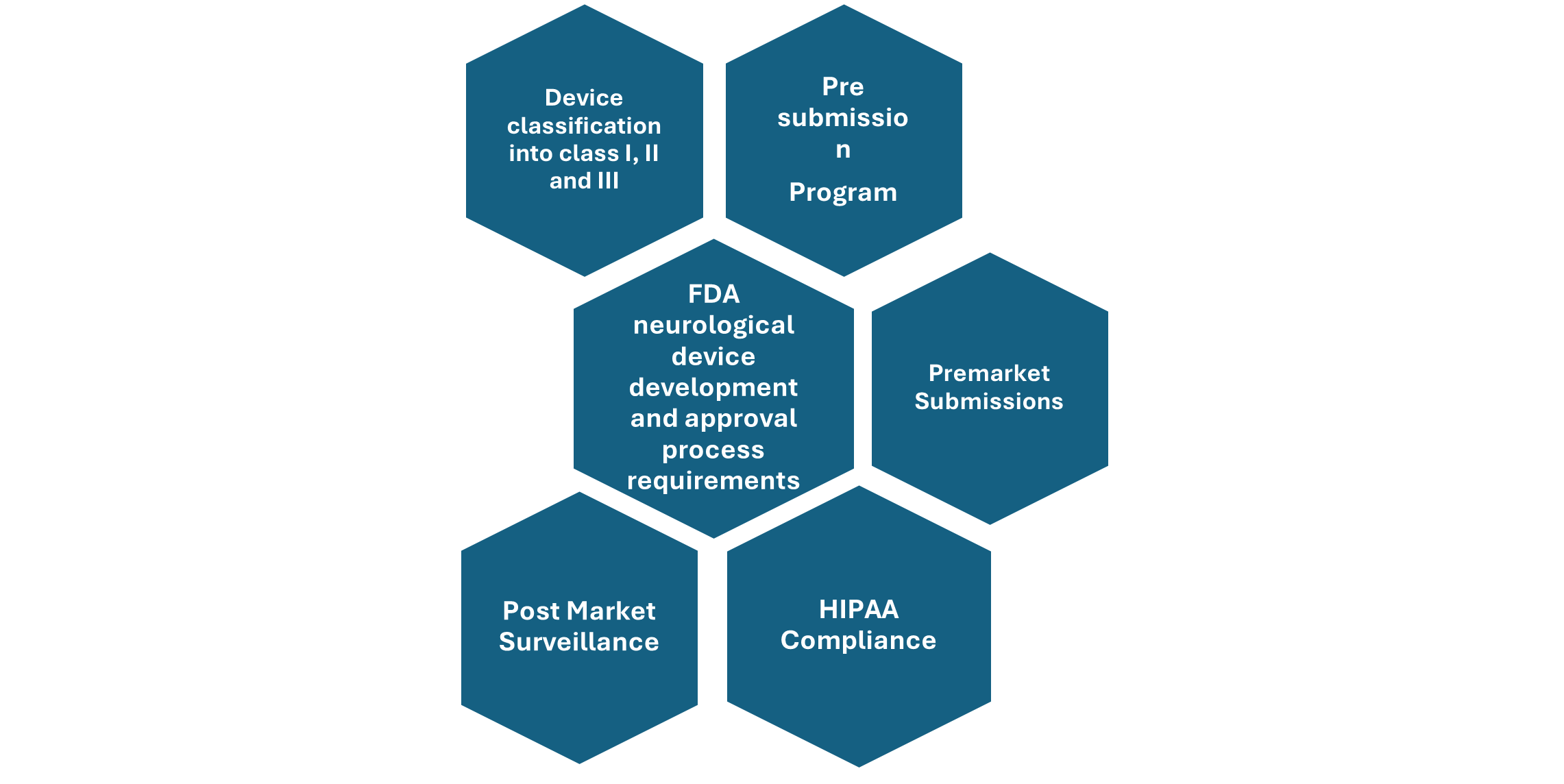
Figure: Overview of FDA neurological Device development and approval process
Neurological device Market Size Overview:
As per MRFR analysis, the Neurology Devices Market Size was estimated at 15.69 (USD Billion) in 2024. The Neurology Devices Market Industry is expected to grow from 16.91 (USD Billion) in 2025 to 33.31 (USD Billion) till 2034, at a CAGR (growth rate) is expected to be around 7.82% during the forecast period (2025 - 2034).
Neurological device Regulatory Landscape:
There are several key regulatory agencies who oversee the approval and monitoring of Neurological device to ensure their safety, efficacy, and quality.
|
Regulatory agencies |
Regulatory Ministry |
|
Federal Food and Drug Administration |
United States: Department of Health and Human Services (HHS) |
|
The Medicines and Healthcare products Regulatory Agency |
United Kingdom: The Medicines and Healthcare products Regulatory Agency (MHRA) under the Department of Health and Social Care (DHSC) |
|
Central Drug Standard Control Organization |
India: The Ministry of Health and Family Welfare |
|
South African Health Products Regulatory Authority (SAHPRA) |
National Department of Health. |
|
Pharmaceuticals and Medical Devices Agency (PMDA) |
Japan: Ministry of Health, Labour and Welfare. |
|
National Medical Products Administration (NMPA) |
China: The Ministry of Health |
|
Health Sciences Authority |
Singapore: The Ministry of Health |
|
European Medicine Agency |
European union |
|
Brazilian Health Regulatory Agency (Anvisa) |
Ministry of Health, part of the Brazilian National Health System (SUS) |
Neurological device Guidelines:
Patients who are diagnosed with neurological disorder like epilepsy, Parkinson’s disease or multiple sclerosis or patients with spinal cord injuries. This treatment option is usually prescribed by the physician when the patient with neurological disorders don’t respond to other medical treatments like drugs or other therapeutics, therefore they specially considered as an effective treatment option for medically resistant neurologic and neuropsychiatric disorders. Use of surgical implants or other invasive devices must be done very carefully and only if required.
Neurological device Classification of the Product:
Neurological device Regulatory Process Overview, By Country:
US Food and drug administration (FDA) regulates neurological devices under center for devices and radiological health (CDRH). These devices fall under medical device regulation and are classified based on the intended use and associated risk.
Key FDA regulations for Neurological Devices:
Device classification- neurological devices are classified as class I which includes devices with least risk to patients, then Class II which comprise devices with moderate risk to the patients, and Class III devices which can pose high risk to the patients utilizing it in the treatment of neurological disorders. Most of these neurological devices are classified under Class II and Class III of medical devices.
Pre-Submission program-The FDA has established a pre-submission program that allows sponsors to interact with FDA staff to identify the most effective regulatory pathway for introducing a medical device to the U.S. market.
The FDA advises sponsors of neurological devices to utilize this pre-submission program to assess whether their planned studies pose significant risks to patients, determine if an Investigational Device Exemption (IDE) is necessary before conducting the study, or to gather additional guidance on the data required for a future marketing submission.
Premarket Requirements- Before marketing a medical device in the United States, several steps must be followed, which include classifying your device, selecting the correct premarket submission, preparing the necessary information for the submission, sending it to the FDA, interacting with FDA during the review, and completing the establishment registration and device listing. The classification of your device determines the type of premarket submission required, and they are as follows:
Premarket notification 510 (k)- Class II devices need premarket Notification 510(K), which demands substantial equivalence evidence of the legally marketed device to prove the safety and efficacy of the new device under the regulation process.
Premarket Approval (PMA)- Class III devices are considered to be life threatening devices and are of high risk, so they undergo strict FDA review during the approval process, these devices are taken for extensive clinical testing and require Premarket Approval (PMA).
Investigational Device Exemption (IDE)- For clinical studies, the FDA's Investigational Device Exemption (IDE) process allows investigational devices to be used to collect data on their safety and effectiveness. High-risk devices require both FDA and Institutional Review Board (IRB) approval before clinical studies can begin. Non-significant risk devices only need IRB approval.
De Novo Grant- The De Novo request offers a marketing pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use, but lack a legally marketed predicate device. This classification is based on a risk-based process.
Humanitarian device exemption (HDE)- A marketing application for a Humanitarian Use Device (HUD) under Section 520(m) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) involves an HDE (Humanitarian Device Exemption). HDE is exempt from the effectiveness requirements of Sections 514 and 515 of the FD&C Act but is subject to certain profit and use restrictions.
A Humanitarian Device Exemption (HDE) is a regulatory pathway for medical devices intended to treat or diagnose diseases affecting no more than 8,000 patients in the U.S. It reduces the regulatory burden on manufacturers by exempting them from the effectiveness requirements of a premarket approval (PMA). To obtain approval, manufacturers submit an HDE application to the FDA, which is similar to a PMA but focuses on safety rather than effectiveness. This pathway encourages the development of devices for small patients. Neurological devices such as deep brain stimulators and aneurysm treatment devices are approved through HDE.
HIPAA (Health Insurance Portability and Accountability Act)- sets national privacy standards to protect patient health information shared among healthcare professionals, medical offices, and hospitals. It ensures patient privacy, data security, and regulates the use of sensitive health information. This regulatory framework also ensures the safety, quality, and efficacy of medical devices, including mobile medical applications.
Post market surveillance- Medical device manufacturers must follow specific regulations once their devices are on the market. This includes mandatory reporting of any incidents where the device may have caused or contributed to a death or serious injury. Postmarket requirements can also involve device tracking, establishment registration, postmarket surveillance studies, and post-approval studies required under section 522 of the Food and Drug Cosmetic Act.
The FDA is encouraging the development of medical device registries to gather real-world evidence on device performance and outcomes. These registries can aid in regulatory decision-making and postmarket surveillance. The FDA collaborates with neurological device manufacturers and professional societies such as NeuroPoint Alliance and NIH (StrokeNet) to develop these registries.
Examples of the certain neurological devices and the regulatory submission they need:
|
Neurological Device |
Class |
Regulatory Pathway |
|
ventricular needles |
Class I |
General Controls |
|
Anvils to form skull plates |
Class I |
General Controls |
|
Neurostimulators |
Class II |
Premarket Notification510 (K) |
|
Aneurysm clips |
Class II |
Premarket Notification510 (K) |
|
Blood clot retrievers |
Class II |
Premarket Notification510 (K) |
|
Deep brain stimulators |
Class III |
Premarket Approval (PMA) |
Neurological device updates:
May 2025, Neuralink, Elon Musk's brain-chip company, has received FDA approval for a new brain implant designed to help individuals with severe speech impairments communicate. This implant, which has been granted the "Breakthrough Device Designation," aims to assist those with conditions such as ALS, stroke, spinal cord injuries, cerebral palsy, and multiple sclerosis. Neuralink is now preparing for human trials and has invited individuals to register through its Patient Registry.
March 2025, TrainHealth announced that its new device, TrainFES Advanced, has received FDA clearance. This device is designed to improve outcomes in neurological rehabilitation by using smart electrical stimulation to move the paralyzed limbs of patients, enabling effective and engaging daily training. The device aims to address the needs of over 8 million people in the U.S. affected by stroke and spinal cord injuries. TrainHealth's CEO, Matias Hosiasson, highlighted that this clearance is a significant step towards transforming neurological rehabilitation. The company has been collaborating with top universities and hospitals globally to advance its technology. According to a study by the University of Illinois Chicago, the device has shown significant improvements in gait and balance, with an 84% adherence rate and up to 40% improvements in motivation and usability.
Neurological device Regulatory Challenges and possible risk in development:
Neurotechnology, like brain-computer interfaces (BCIs), faces several regulatory challenges and risks. These devices raise ethical issues about privacy, identity, and consent. BCIs collect a lot of neural data, so strong protections against unauthorized access are needed to keep this information safe. Neural data can reveal private thoughts and memories, Brain implants might change how people think and behave, raising questions about personal identity and authenticity.
Human trials of BCIs require clear informed consent processes, ensuring participants understand the risks and outcomes, including short-term surgical risks and long-term cognitive effects. The newness of neurotechnology makes it hard to predict all consequences, so transparency and ongoing consent is very important. Invasive BCIs carry surgical risks like infection or hemorrhage, and long-term reliability is a concern as implants may degrade over time. These challenges highlight the need for new regulatory frameworks and ethical guidelines to ensure the safe and responsible development of neurotechnology.
Complying with HIPAA to overcome all the ethical and privacy related issues is utmost important and challenging. Overcoming technological challenges related to the device during development process to develop the product meeting the regulatory standards, further add to the challenges.
Complex approval pathways for neurology devices, especially those which are implantable or interact directly with the brain, falling under class II and class III medical devices, requiring premarket notification or premarket approval or de novo classification or CE marking under EMA demanding substantial equivalence evidence and extensive testing, so the whole process can be costly and time consuming. If any adverse event is reported under MDR Post market surveillance it can lead to product recall and huge loss for the company.
Neurological device Competitive Landscape Dashboard:
Companies With Marketed Neurological devices:
- Johnson and Johnson
- Gore & Associates
- Braun Mesungen AG
- Jude Medical Inc.
- Integra LifeSciences Holdings
- Esaote
- Boston Scientific
- Stryker Corporation
- Medtronic PLC (Ireland)
- Rapid Medical
- Acandis GmbH
- MicroPort Scientific Corporation
- Perflow Medical
- Lepu Medical, among others
Regulatory Landscape - Table of Content
Table of contents will appear here once available.
Customer Stories

“This is really good guys. Excellent work on a tight deadline. I will continue to use you going forward and recommend you to others. Nice job”

“Thanks. It’s been a pleasure working with you, please use me as reference with any other Intel employees.”

“Thanks for sending the report it gives us a good global view of the Betaïne market.”

“Thank you, this will be very helpful for OQS.”

“We found the report very insightful! we found your research firm very helpful. I'm sending this email to secure our future business.”

“I am very pleased with how market segments have been defined in a relevant way for my purposes (such as "Portable Freezers & refrigerators" and "last-mile"). In general the report is well structured. Thanks very much for your efforts.”

“I have been reading the first document or the study, ,the Global HVAC and FP market report 2021 till 2026. Must say, good info! I have not gone in depth at all parts, but got a good indication of the data inside!”

“We got the report in time, we really thank you for your support in this process. I also thank to all of your team as they did a great job.”
