
Regulatory Landscape - Overview
Cell and gene therapy Regulatory Landscape: Product Overview
Cell and gene therapy are advanced medical approaches that involve the introduction, manipulation, or modification of genetic material within cells to treat or prevent diseases. These therapies aim to correct genetic defects, enhance the body's ability to fight diseases, or replace dysfunctional cells with healthy ones.
The Centre for Biologics Evaluation and Research (CBER) division of Food and drug administration (FDA) is responsible for the regulation of cellular therapy products, human gene therapy products, and certain devices related to cell and gene therapy. They use both the Public Health Service Act and the Federal Food Drug and Cosmetic Act for regulation to ensure that the novel cellular and gene therapy products manufactured are of high quality, safe and effective.
Cell and gene therapy types
Cell Therapy is segmented into Autologous and Allogeneic, whereas Gene Therapy is segmented into two principal classes: Germline gene therapy (GGT), and somatic cell gene therapy (SCGT), somatic gene therapy is further classified into Ex vivo and In vivo types.
Cell and gene therapy Administration Overview:
Cell therapy
In autologous cell therapy, patients own cells are extracted, manipulated, and reinfused into patients own body. Example includes CAR-T therapy, wherein there is a collection of T cells from cancer patients’ body, and they are genetically modified to target cancer cells and reinfused into the patient’s body. This reduces the risk of immune rejection as patients own cells are utilised for the treatment.
In allogenic therapy, cells are taken from donor, who is related or unrelated to the patient. Example include hematopoietic stem cells transplants for leukemia treatment, wherein patients diseased bone marrow is replaced by utilising healthy donor stem cells. There is higher risk of immune rejection and complications like graft versus host disease (GVHD) can occur in patient.
Gene therapy
In germ line gene therapy, there is a manipulation in reproductive cell line and somatic gene therapy involves correction of genetic anomalies in non-reproductive cells.
Under somatic gene therapy, in Ex vivo type, there is removal of cells from the body, followed by incubation with a vector, and then engineered gene cells are reinfused into the body, usually this is done with blood cells, as they are the easier to be removed and then reinfuse. In in vivo type, vector is injected directly into the bloodstream.
Cell and gene therapy Applications:
Allogenic Stem Cells Based Gene Therapy for Crohn’s Disease
biotechnology companies in Belgium and Japan, namely, TiGenix and Takeda, have developed the treatment of perianal fistulas, which is the serious manifestations of Crohn’s disease. It is an an autoimmune disease concerning the large and small intestine, these patients require specialized diets and medicines. Cx601 – the gene therapy use specialized adipose tissue cell constructs – which has ability to reduce host cell deduction and stimulate immunomodulatory effects of T cells. This unique expression of adipose-based stem cells enabled the generation of normal regulatory T cells that prevented the formation of perianal fistulas.
Gene therapy for neurological disorders
Gene therapy is used for the treatment of neurological disorder, involves delivery of therapeutic genes to neurons, for correction of mutations or restoring essential protein function.
Example: Zolgensma, an AAV9-based gene therapy, approved by FDA, utilised for treatment of spinal muscular atrophy (SMA), severe neurodegenerative disease, caused due to mutations in SMN1 gene, and causing deficiency of survival motor neuron protein. Zolgensma delivers functional SMN1 copy to motor neuron, helping to restore SMN protein level and overcome the disease.
Gene therapies for eye diseases
Interesting application of gene therapy can be seen in addressing ophthalmological disorders, like inherited retinal diseases (IRDs) which can cause gradual vision loss. Gene therapy delivers functional genes directly to retinal cells, which helps in correcting the genetic mutations which cause the disease.
Example: First FDA approved Spark Therapeutics’ Luxturna for an inherited retinal disease. It is designed for patients having RPE65 mutation-associated retinal dystrophy, which can result in severe vision impairment or blindness. Therapy uses an AAV vector for delivering a healthy copy of the RPE65 gene directly to retinal cells through subretinal injection, helping to treat the disease.
Gene therapy for cancer
Gene therapy for oncology focuses on manipulation of genetic material to restore normal cellular functions, silence oncogenes, or increase sensitise cancer cells for other treatments.
Examples: utilising of gene silencing techniques for targeting oncogenes, approaches like antisense oligonucleotides (ASOs) and RNA interference (RNAi) are used for downregulation of harmful genes like KRAS, which is frequently mutated in pancreatic and colorectal cancers. CAR-T therapy, and cell-based gene therapy, involves genetic modification of patients own T cells for enhancing ability of identification and elimination of cancers, CAR-T therapies which got regulatory approval includes Yescarta and Kymriah, targeting CD19 protein, expressed on the surface of B-cell malignancies.
Cell and gene therapy Product Development Steps:
FDA Approval Process for Cell and gene therapy Product
The FDA has formed four methods to speed up the development of new drugs: Fast Track, Breakthrough Therapy, Accelerated Approval (AA), and Priority Review.
Few recent fast track and Breakthrough Designation granted by FDA to Cell and Gene Therapies include the following:
- February 2025, Bayer AG and BlueRock Therapeutics LP, has announced U.S. FDA grant of fast-track designation OpCT-001, an investigational induced pluripotent stem cell (iPSC)-derived cell therapy for the treatment of primary photoreceptor diseases.
- 2023, U.S. FDA granted fast track designation and breakthrough designation to CG Oncology’s cretostimogene grenadenorepvec for treating patients with Bacillus Calmette-Guerin (BCG)-unresponsive non-muscle invasive bladder cancer (NMIBC).
Product development steps include:
Preclinical
- Interact meeting
- Pre IND meeting
IND submission
Clinical trials
- Phase 1 – safety and dosage evaluation
- Phase 2- efficacy assessment and further safety monitoring
- Phase 3- large scale trials to ensure efficacy
Pre BLA meeting
Marketing Application
- BLA
Post marketing
Cell and gene therapy Market Size Overview:
As per MRFR analysis, the Cell and gene therapy Market Size was estimated at 15,018.57 (USD Million) in 2024. The Cell and gene therapy Market Industry is expected to grow from 17,639.31 (USD Million) in 2025 to 67,891.47 (USD Million) till 2032, at a CAGR (growth rate) is expected to be around 18.12% during the forecast period (2023-2032). Growing incidences of cancer and rare disorders and increasing approval for gene and cell therapy products are the key market drivers to enhancing the growth of the market.
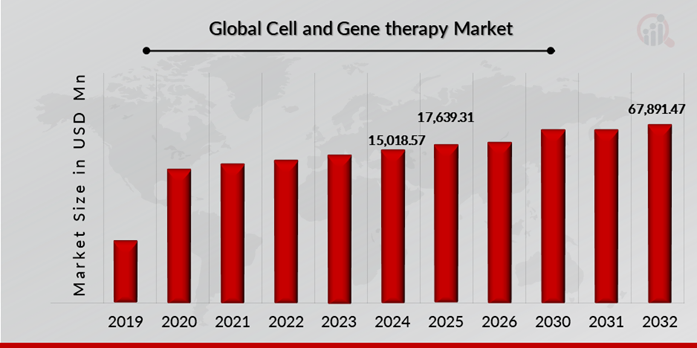
Source: The Secondary Research, Primary Research, MRFR Database and Analyst Review
Cell and gene therapy Regulatory Landscape:
There are several key regulatory agencies who oversee the approval and monitoring of Cell and gene therapy to ensure their safety, efficacy, and quality.
| Regulatory agencies | Regulatory Ministry |
| Federal Food and Drug Administration | United States: Department of Health and Human Services (HHS) |
| The Medicines and Healthcare products Regulatory Agency | United Kingdom: The Medicines and Healthcare products Regulatory Agency (MHRA) under the Department of Health and Social Care (DHSC) |
| Central Drug Standard Control Organization | India: The Ministry of Health and Family Welfare |
| Health Canada | Canada: The Ministry of Health |
| Pharmaceuticals and Medical Devices Agency (PMDA) | Japan: Ministry of Health, Labour and Welfare. |
| National Medical Products Administration (NMPA) | China: The Ministry of Health |
| Health Sciences Authority | Singapore: The Ministry of Health |
| European Medicine Agency | European union |
| Therapeutic Goods Administration (TGA) | Commonwealth of Australia |
Cell and gene therapy Guidelines:
Eligibility: Cell and Gene therapy are mostly administered to the patients suffering with serious genetic disorders, cancers or chronic diseases, which can’t be cured with conventional treatments, cost and availability of the treatment play major role in access to these treatments. Eligibility depends on disease type, age of the patient and patient condition.
There are several controversies regarding the practice of cell and gene therapy. Manipulation of germline cells at gene level can permanently eradiate certain hereditary disorders, but major ethical issues like eugenics, enhancement, mosaicism and transmission of undesirable traits or side effects to patients descendants is currently hampering its use or strictly regulated in most countries, and leaving only somatic gene therapy to use in therapeutics.
Cell and gene therapy Classification of the Product:
Cell and gene therapy Regulatory Process Overview, By Country:
FDA Approval Process for Cell and gene therapy
FDA has issued guidance prepared by Office of Therapeutic Products (OTP), under center for biologics evaluation and research (CBER).
OTP looks after the development of a variety of products which includes purified, recombinant protein for hematology, gene therapies, cell therapies, human tissue products, therapeutic tissue engineered products any many more.
Key steps in cell and gene therapy (CGT) development and approval include following;
Preclinical step
When developing a novel product, a sponsor may want to obtain OTP’s advice on the data needed to support the submission of an Investigational New Drug application (IND). INTERACT and Pre-IND meetings may be held at these early stages of product development.
Interact meeting is a Regulatory Advice for initial Targeted Engagement for on CBER/CDER Products, this meeting is a chance for sponsors developing novel therapies to ask for feedback at an early stage of development.
Pre-IND Meeting its primary goal is to review and get feedback on the preclinical studies design, the design of the initial IND study, and product manufacturing and quality controls needed to initiate human studies.
Investigational new drug (IND) submission, it contains details of drugs potential and safety data (drug substance, drug product characterization), needs to be submitted before human clinical trials.
3 stages of 30 days IND review process of FDA.
Stage 1 – FDA document control center (DCC) processes the IND submission, Office of Therapeutic Products (OTP) confirms the submission is in correct office, Regulatory Project Manager (RPM) performs administrative review to ensure completeness. If it is complete sponsor receive an acknowledgement letter.
Stage 2 – scientific review and assignment, reviewers are assigned from disciplines like CMC (chemistry, manufacturing, controls), pharmacology/ toxicity (PT) and clinical, additional experts are also assigned if required.
Stage 3- FDA issues, either safe to proceed decision or clinical hold if any concerns related to products are determined.
Clinical trials
In this phase the end of phase 1 and 2 meetings and pre BLA meetings are undertaken ; End of Phase (EOP) Meeting purpose is to review the overall of the development program status, including available clinical safety and efficacy data, also include addressing all important questions or issues for all disciplines [e.g., Chemistry, Manufacturing, and Controls (CMC), pharmacology/toxicology (P/T), and clinical] that are required for the further development of the product.
Sponsor should schedule a pre BLA meeting with OTP review team. This meeting will ensure that BLA submission includes all necessary data, which in turn will reduce delays by discussion of key issues before submission. Meeting request need to be submitted 4 months before BLA submission.
FDA expedited programs for CGT products
Rolling review means allows FDA to consider reviewing portions of a BLA before submission of complete BLA by sponsor. A drug can be granted rolling review if certain criteria are met, and it has it has Fast Track, Breakthrough Therapy, Regenerative Medicine Advanced Therapy (RMAT) designation, these are expedited programs of FDA which are applicable to advanced therapies such as cellular and gene therapy products with the intension to treat, modify, reverse, or cure a serious condition. Therapeutics developed under BT and RMAT will follow more intensive guidance on development programs with frequent interactions OTP.
Market application
Biological License Application (BLA) submission and review
This submission must include final clinical trial data, covering safety and efficacy details, validated potency and quality testing results, manufacturing process validation, with current good manufacturing practice (CGMP) compliance.
Post marketing
Post marketing surveillance for long term safety monitoring through required studies, and adverse event reporting, if company or customers finds any unexpected safety issues with the therapeutics developed.
Selection of animal models for preclinical study- FDA recommendations
Key considerations while selecting animal models for non-clinical pharmacology and toxicology study;
- Whether investigational cell and gene therapy products are pharmacologically active in species.
- Physiological and anatomical compatibility between animal and humans for route of administration and target anatomic sites.
- Sensitivity of selected species to potential toxicities for the product.
Special considerations for Cell and Gene Therapy
- Ability of species to support survival and engraftment of cell therapy products
- Susceptibility of species to vector, vector transduction profile
- Pharmacological response to vector and expressed transgene
Alternative approaches when animal models of disease are not available
- In vitro studies using direct human cells
- Use of in silico (computational) models
- in vivo studies using an analogous animal product
- relevant nonclinical or clinical data from studies evaluating a related product
or indication.
Major Cell and Gene Therapy Approved Products
| Company Name (Headquarter) | Approval Date | Brand Name | Indication | Therapy Type |
| Orchard Therapeutics (Japan) | March 2024 | Lenmeldy | Metachromatic Leukodystrophy | Gene Therapy |
| Janssen Biotech, Inc. (Belgium) | February 2024 | CARVYKTI | Earlier Lines of Relapsed and Refractory Multiple Myeloma | Cell Therapy |
| Ferring Pharmaceuticals (Switzerland) | December 2022 | Adstiladrin | Bacillus Calmette-Guérin (BCG)unresponsive non-muscle invasive bladder cancer (NMIBC) | Gene Therapy |
| Bristol Myers Squibb (US) and Juno Therapeutics, Inc. (US) | June 2022 | Breyanzi | Relapsed or Refractory Large B-cell Lymphoma | Cell Therapy |
Source: Company News, US Food Drug and Administration, MRFR Analysis
Cell and gene therapy Regulatory Updates:
The FDA has issued 2 final documents containing guidance for the biotherapy community, this finalizes draft guidance’s of the same titles.
First guidance for Human Gene Therapy Products Incorporating Human Genome Editing includes recommendatory guidelines for sponsors of human gene therapy products, incorporating genome editing (GE) of somatic cells of humans. Contains information that sponsors should submit Investigational New Drug application (IND) for accessing the safety, efficacy and quality of investigational gene editing product, as per the requirement of federal regulations, and this include information of product design, manufacturing and testing, nonclinical safety assessment and clinical trial design
Second guidance for Considerations for the Development of Chimeric Antigen Receptor (CAR) T-Cell Products, FDA suggest recommendatory guidelines to sponsors developing CAR T cell products regarding chemistry, manufacturing and control (CMC), pharmacology and toxicology and design of clinical studies for oncology indications, even it includes specific recommendations for autologous or allogenic CAR-T cell products and analytical comparability studies for CAR-T cell products.
Cell and gene therapy Regulatory Challenges:
Regulatory challenges company developer face includes meeting quality, safety and efficacy of the therapeutics, correct estimation of dosage, development of manufacturing processes, invasive route of administration, small patient population for the rare disease applications, lack in clinical endpoints. Cell and gene therapies have potential risk of long-term immunogenicity and tumorigenicity or loss of expression over time.
Clinical Trial Failure Associated with Gene and Cell Therapy
Clinical trial failures associated with gene and cell therapy have a significant impact on the market's growth trajectory. Despite promising preclinical data, some therapies may fail during clinical trials due to a variety of factors, including unexpected side effects, insufficient efficacy, or difficulties in effectively delivering the therapy to target tissues. These setbacks not only result in significant financial losses for companies, but they also undermine investor confidence in the viability of gene and cell therapy as a treatment modality. For instance, in October 2021, Novartis AG (Switzerland) terminated its Rett syndrome gene replacement program, OAV201, after pre-clinical data did not support a path forward to human clinical trials. Moreover, in September 2023, Taysha Gene Therapies (US) revealed that it has discontinued TSHA-120 development for gene therapy. This is because the FDA requested changes to the Phase I/II study designs for TSHA-120 in giant axonal neuropathy. Furthermore, high-profile clinical trial failures can harm public perception and increase regulatory scrutiny, slowing the approval process for other therapies under development.
Possible Risk in development of Cell and gene therapy:
Capacity constraints in manufacturing, high cost of the goods, raw materials including cell lines, culture media, plasmids and many more, stringent regulatory requirements further increase quality assurance expenses, long lead time, significant upfront investment requirements, delay in clinical trials can result in longer time to get launched for the therapeutic to the market and can hamper market growth.
Most countries don’t have research and medical infrastructure required for developing regulatory framework to support timely and efficient development of gene therapies, leaving with limited access to transformative treatment options for many patients.
Overcoming Cell and gene therapy product development challenges:
Different countries have their own regulatory guidelines for approval of therapeutics used in various disease treatment, which can create challenge for global therapeutics approval, successful navigation of these global regulatory complexities can be done by adhering to the international council of harmonization (ICH) guidelines, ensuring compliance across multiple regions with different regulatory requirements, allowing companies to follow common sets of standards. Many regulatory bodies like FDA and EMA use ICH guidelines to align their therapeutic approval pathways to meet global standards, for smoother global approvals.
For preventing immunogenicity issues in cell and gene therapy we can avoid the use of immunogenic epitopes in developing the therapeutics.
Cell and gene therapy Competitive Landscape Dashboard:
Companies With Marketed Cell and gene therapy
- Novartis AG
- Bristol-Myers Squibb Company
- Amgen Inc.
- Aurion Biotech
- Sarepta Therapeutics, Inc.
- Ferring B.V.
- Sibiono GeneTech Co. Ltd., Shenzhen (Sibiono)
- Janssen Global Services
- bluebird Bio, Inc.
- Kite Pharmaceuticals, Inc
Regulatory Landscape - Table of Content
Table of contents will appear here once available.
Customer Stories

“This is really good guys. Excellent work on a tight deadline. I will continue to use you going forward and recommend you to others. Nice job”

“Thanks. It’s been a pleasure working with you, please use me as reference with any other Intel employees.”

“Thanks for sending the report it gives us a good global view of the Betaïne market.”

“Thank you, this will be very helpful for OQS.”

“We found the report very insightful! we found your research firm very helpful. I'm sending this email to secure our future business.”

“I am very pleased with how market segments have been defined in a relevant way for my purposes (such as "Portable Freezers & refrigerators" and "last-mile"). In general the report is well structured. Thanks very much for your efforts.”

“I have been reading the first document or the study, ,the Global HVAC and FP market report 2021 till 2026. Must say, good info! I have not gone in depth at all parts, but got a good indication of the data inside!”

“We got the report in time, we really thank you for your support in this process. I also thank to all of your team as they did a great job.”
