
Regulatory Landscape - Overview
Biosimilars Regulatory Landscape: Product Overview
Biosimilars are biologic medical products that are highly like an already approved reference biologic drug in terms of safety, efficacy, and quality. Unlike traditional small-molecule drugs, which have simple chemical structures and can be exactly replicated as generics, biologics are large, complex molecules derived from living cells. Due to this complexity, biosimilars are not identical copies but must demonstrate no clinically meaningful differences from their reference products.
Biosimilars play a crucial role in expanding patient access to life-saving treatments, particularly for chronic and life-threatening diseases like cancer, autoimmune disorders, and diabetes. By introducing competition in the biologics market, they help lower healthcare costs while maintaining high treatment standards. As more patents for original biologics expire, the biosimilars market continues to grow, offering healthcare systems worldwide more affordable alternatives without compromising quality or therapeutic benefits.
FDA and EMA, require extensive analytical, preclinical, and clinical studies to confirm biosimilarity before approval. Biologics price competition and innovation act of 2009 (BPCI Act) has developed regulatory approval pathway for providing help to patients with greater access to safe and effective biological products. FDAs biosimilar action plan (BAP) encourages innovation and competition for biologics and facilitate the development of safe and effective biosimilar and interchangeable biosimilar products with low cost, helping to reduce time and cost of development without compromising safety and effectiveness.
Biosimilars types
Biosimilars of various types used for the treatment of various diseases including monoclonal antibodies, insulin, Erythropoietin, Growth Hormones.
List of biosimilars approved by FDA and EMA falling in above mentioned categories include;
| Monoclonal Antibodies | Insulin | Erythropoietin | Growth hormones |
| • Adalimumab • Infliximab • Rituximab • Bevacizumab • Trastuzumab • Ustekinumab • Tocilizumab • Aflibercept • Dupilumab • Denosumab | Merilog Rezvoglar Semglee | Retacrit Binocrit silapo | Omnitrope (somatropin) Valtropin Sogroya |
Source-MRFR analyses
Biosimilars Applications
Biosimilars offer a more affordable option compared to their reference biologics, often at a fraction of the cost. By providing similar therapeutic benefits, biosimilars are increasingly being adopted across various therapeutic areas, including oncology, autoimmune diseases, and diabetes. Regulatory support, coupled with advancements in biomanufacturing technologies, has further accelerated their development and market penetration. As healthcare systems prioritize cost containment without sacrificing patient outcomes, biosimilars are poised to play a pivotal role in shaping a more sustainable and accessible healthcare landscape.
Biosimilar monoclonal antibodies are utilised for treatment of various health diseases like; in oncology they target specific antigen on cancer cells, aiding treatment of various malignancies, in autoimmune disorders they modulate immune responses providing therapeutic options for conditions like rheumatoid arthritis and inflammatory bowl disease.
Growth hormones are used for treating growth hormone deficiencies, aiding normal growth, to treat metabolic imbalances, improving quality of life in patients lacking adequate growth hormone levels.
Insulin biosimilars are developed for treatment of diabetes, for instance Merilog, which assist in controlling blood glucose levels in diabetic patients.
Biosimilar Product Launch:
May 2024: Teva Pharmaceuticals and Alvotech launched of SIMLANDI (adalimumab-ryvk) injection in the U.S. as the first interchangeable, high-concentration, citrate-free biosimilar to Humira. This biosimilar is designed to treat a range of conditions, including adult rheumatoid arthritis, juvenile idiopathic arthritis, adult psoriatic arthritis, adult ankylosing spondylitis, Crohn's disease, adult ulcerative colitis, adult plaque psoriasis, adult hidradenitis suppurativa, and adult uveitis.
October 2023: The FDA approved Celltrion USA's Zymfentra (infliximab-dyyb , the world’s first and only subcutaneous infliximab product. Approved as a novel drug, Zymfentra is developed based on Remicade, the reference intravenous infliximab. It is specifically approved for the treatment of adults with moderate to severe inflammatory bowel disease IB, which includes both ulcerative colitis (UC) and crohn’s disease (CD) intravenously administered infliximab product.
December 2019: Daiichi Sankyo has announced the launch of its bevacizumab biosimilar for intravenous drip infusions in Japan. The product, developed in collaboration with Amgen, is available in 100 mg and 400 mg doses. It is a biosimilar to the anti-VEGF humanized monoclonal antibody bevacizumab, approved in Japan on September 20, 2019, for treating unresectable advanced or recurrent colorectal cancer. This launch marks a significant step in providing cost-effective cancer treatment options in Japan.
Biosimilar Product development
FDA do careful review of data, studies, different tests, Medication quality checking during development and production, patient safety reports review made to FDA, to ensure safe and effective development of biosimilars as their original product. Even monitor the safety and efficacy after their approval. Office of Therapeutic Biologics and Biosimilars (OTBB) under FDA, is responsible for the development and approval of safe and effective biosimilar and interchangeable products.
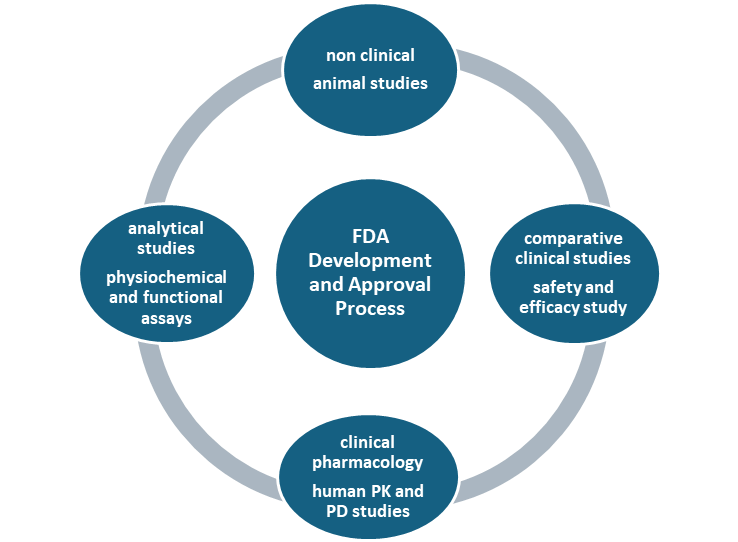
Fig-Biosimilar Product Development Overview
Biosimilars Market Size Overview:
As per MRFR analysis, the Biosimilars Market Size was estimated at 32,729.17 (USD Million) in 2024. The Biosimilars Market Industry is expected to grow from 37,768.39 (USD Million) in 2025 to 98,354.07 (USD Million) till 2032, at a CAGR (growth rate) is expected to be around 14.74% during the forecast period (2024 - 2032). Growing incidences of cancer and rare disorders, increasing launch of biosimilars, rising healthcare costs are the key market drivers enhancing the growth of the market.
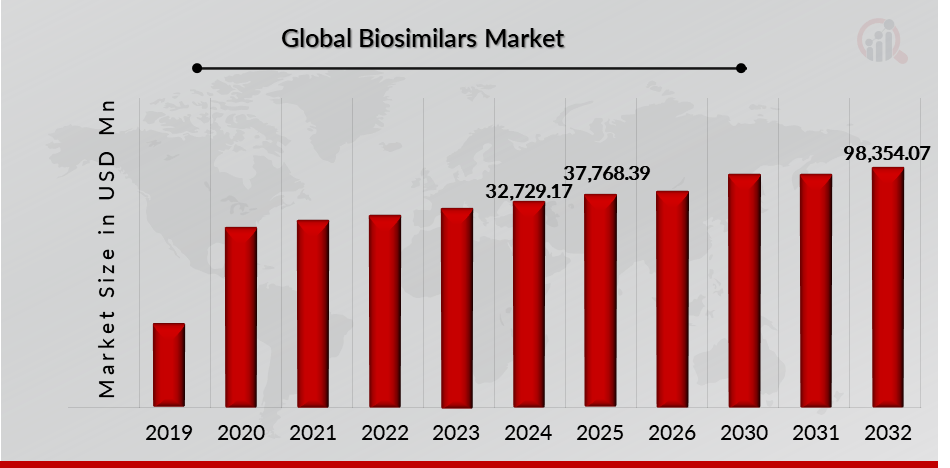
Source: Secondary Research, Primary Research, MRFR Database and Analyst Review
Biosimilars Regulatory Landscape:
There are several key regulatory agencies oversee the approval and monitoring of Biosimilars to ensure their safety, efficacy, and quality.
| Regulatory agencies | Regulatory Ministry |
| Federal Food and Drug Administration | United States: Department of Health and Human Services (HHS) |
| The Medicines and Healthcare products Regulatory Agency | United Kingdom: The Medicines and Healthcare products Regulatory Agency (MHRA) under the Department of Health and Social Care (DHSC) |
| Central Drug Standard Control Organization | India: The Ministry of Health and Family Welfare |
| Health Canada | Canada: The Ministry of Health |
| Pharmaceuticals and Medical Devices Agency (PMDA) | Japan: Ministry of Health, Labour and Welfare. |
| National Medical Products Administration (NMPA) | China: The Ministry of Health |
| Health Sciences Authority | Singapore: The Ministry of Health |
| European Medicine Agency | European union |
| Therapeutic Goods Administration (TGA) | Commonwealth of Australia |
Eligibility: biosimilars are used as safe and effective treatment options for many diseases treatments like chronic skin and bowel diseases like psoriasis, irritable bowel syndrome, Crohn’s disease and colitis), arthritis, kidney conditions, and cancer.
Patients can switch to biosimilar due to a change in insurance coverage or to save money spent on treatment, as biosimilars are cost affordable. The low cost of biosimilars is covered by insurance companies, offering patients additional treatment options.
Biosimilars Classification of the Product:
Biosimilars Regulatory Process Overview, By Country:
FDA Approval Process for Biosimilars
There is a rigorous evaluation of biological products including biosimilars and interchangeable biosimilars, for ensuring safety, quality, potency, and efficacy. An abbreviated licensure pathway is created in section 351(k) of the Public Health Service Act (PHS Act) by BPCI Act, for all the biological products showing the potential of biosimilarity, interchangeable with an FDA licensed biological reference product.
Centre for Drug Evaluation and Research (CDER) and the Centre for Biologics Evaluation and Research (CBER) at the Food and Drug Administration (FDA or the Agency) has even issued a regulatory guidance to be followed during development and approval process for industries.
Regulatory steps to be followed by Industries for development of biosimilars, given by FDA
Standalone application- this is for approval of reference product and must contain all data and information necessary to demonstrate the product’s safety and effectiveness
Analytical studies – company developing biosimilars must give comparative analytical data proving that proposed biosimilar is highly similar in structure and function to reference product, even do evaluation of impact of any differences if identified.
Animal studies- this involves pharmacological and toxicity study which provide additional support for biosimilarity.
Investigational new drug application- A sponsor may submit a single IND application for a development program that is intended to support licensure of a proposed product under section 351(k) of the PHS Act and includes use of a non-U.S.-licensed product.
If a non-U.S. licensed comparator is used, sponsor must provide a scientific bridge and scientific justification for the relevance of these comparative data to an assessment of biosimilarity to the U.S.-licensed reference product. IND submission should include chemistry, manufacturing and controls (CMC) information required by 21 CFR 312.23. FDA has idea that obtaining all CMC information required by 21 CFR 312.23 for a non-U.S.-licensed comparator product for which it is not the manufacturer. In this case, the sponsor can request FDA to waive the regulatory requirements related to CMC information on the non-U.S.-licensed comparator product.
Clinical pharmacological study- Clinical study aim at addressing if any potential residual uncertainty is present, ensuring that no clinically meaningful differences are present between proposed biosimilar and reference product in all terms like safety, purity, efficacy. Clinical study nature depends on extent of residual uncertainty identified about biosimilarity between the therapeutics after the analytical and in vitro characterisation. This study involves evaluation of pharmacokinetics, pharmacodynamics and immunogenicity assessment to evaluate a patient’s immune response to the biosimilar.
Additional clinical studies- at least one clinical study is required to evaluate immunogenicity, safety, purity, and potency. sometimes conducted after the completion of other studies to address any remaining uncertainty about whether the proposed biosimilar has no clinically meaningful differences from the reference product.
Extrapolation- applicants may seek approval of biosimilar under section 351 (k) of PHS act for multiple indications using scientific justification.
Interchangeability requirements- FDA ask applicants seeking to get interchangeability designation, will need to submit data and information, supporting to showing that the proposed interchangeable product can be expected to produce the same clinical result as reference product
Submission of biologics license application (BLA) under 351 (k)- A BLA submitted under section 351(k) (a “351(k) BLA”) must contain, among other things, information demonstrating that the biological product is biosimilar to a reference product based upon data derived from analytical studies, animal studies, and a clinical study or studies.
FDA review- Biosimilars are evaluated for approval based on all the evidence presented by the manufacturer. The abbreviated approval process maintains the same high approval standards that are applied to all biologics, and FDA’s rigorous standards help to ensure that all approved biosimilars are as safe and effective as their reference products.
Post market surveillance: reporting of any adverse events after administration of approved biosimilar.
Purple book database – has information of all biological products including biosimilars, interchangeable products and their reference products, with granted approval and regulated by CDER and CBER under FDA.
Biosimilar products approved by FDA
| Biosimilar name | Manufacturer | Approval | Reference product |
| Avtozma (tocilizumab-anoh) | CELLTRION INC | January 2025 | Actemra (tocilizumab) |
| Ahzantive (aflibercept-mrbb) | Formycon/Klinge Pharma | June 2024 | Eylea (aflibercept) |
| Bkemv (eculizumab-aeeb) | Amgen | May 2024 | Avastin (bevacizumab) |
| Avzivi (bevacizumab-tnjn) | Bio-Thera solutions, marketed by Sandoz | December 2023 | Avastin (bevacizumab) |
FDA Updates on biosimilars
February 2025, first rapid acting Insulin Biosimilar Product is approved by FDA for treatment of Diabetes, agency is putting efforts for providing increased access to insulin treatment options. The FDA has granted approval of Merilog to Sanofi-Aventis U.S. LLC.
U.S. FDA has approved Merilog (insulin aspart-szjj), which show biosimilarity to Novolog (insulin apart), improving glycaemic control for treatment of diabetes mellitus in adult and paediatric patients. It is a rapid acting insulin which helps to lower mealtime blood sugar spikes to improve control of blood sugar in people with diabetes. 3 ml single patient use prefilled pen and 10 ml multiple dose vial are both approved for use.
FDA approved Merilog as third insulin biosimilar product, which joins two long-acting insulin biosimilar products approved by FDA in 2021. Merilog is administered within 5- 10 minutes before taking meal. It is even administered subcutaneously by injection to stomach, buttocks, thighs or upper arms. Dosing is adjusted based on need of particular patient. It may cause few side effects like severe allergic reactions, hypoglycaemia, hypokalemia, weight gain and swelling problems.
Biosimilars Regulatory challenges:
Regulatory and approval barriers are significant impediments to the growth of the global biosimilars market, despite its potential to provide cost-effective alternatives to biologic drugs. The development of biosimilars requires rigorous and costly processes to demonstrate similarity to reference biologics in terms of safety, efficacy, and quality. According to a report by the Generics and Biosimilars Initiative, the average cost of developing a biosimilar range from 100 million to 300 million, with clinical trials alone accounting for a substantial portion of this expense. Additionally, regulatory requirements vary widely across regions, creating inconsistencies and delays.
For example, while the FDA and EMA have established relatively streamlined pathways, other regions, such as parts of Asia and Latin America, lack clear or harmonized guidelines, leading to prolonged approval timelines. A 2021 study by the RAND Corporation found that biosimilars in the U.S. take an average of 7 to 8 years to reach the market after patent expiration, compared to 3 to 5 years for traditional generics. Furthermore, legal challenges and patent litigations from originator companies often delay market entry, with over 70% of biosimilar launches in the U.S. facing such disputes, as reported by the Association for Accessible Medicines. These barriers not only increase development costs but also limit the timely availability of biosimilars, restraining the global market's growth despite the rising demand for affordable biologic therapies.
Main issue of biosimilar entry to market of US is regulatory exclusivity, regulatory biosimilarity, requirements for interchangeability and furthermore patent exclusivity periods. Even if primary biological patent has expired, launch of affordable biosimilar in market is delayed due to presence of active secondary patent. Secondary patents have given to new formulation, manufacturing processes, novel therapeutics application of the existing active ingredients, and new combinations of known active ingredients. For life cycle management strategy formulation patent is important to retain market exclusivity after expiry of original patent.
Risk in development of Brachytherapy treatment options:
Limited reimbursement and market access policies are playing a pivotal role in driving the growth of the global biosimilar market. As healthcare systems worldwide face increasing cost pressures, biosimilars—which are highly similar to already approved biologic drugs but offered at lower prices—are becoming a preferred alternative to expensive originator biologics. Limited reimbursement policies, which often restrict coverage to the most cost-effective treatments, are encouraging the adoption of biosimilars by making them more financially accessible to patients and healthcare providers.
Additionally, streamlined market access pathways, such as expedited regulatory approvals and supportive frameworks in regions like Europe and the U.S., are further accelerating the entry of biosimilars into the market. This combination of cost containment strategies and favorable regulatory environments is fostering competition, reducing healthcare expenditures, and expanding patient access to essential therapies, thereby fueling the rapid growth of the global biosimilar market.
Even if biosimilars gives prevention and treatment options for many diseases, along with less side effects, low socioeconomic status countries can’t get access to health services due to higher financial burden and lack of accessibility.
Biosimilars Competitive Landscape Dashboard:
Companies With Marketed Biosimilars Products:
-
Novartis AG
-
Pfizer lnc.
-
Amgen, lnc.
-
Biogen
-
Eli Lily and Company
-
Teva Pharmaceuticals
-
Samsung Bioepis Co., Ltd
-
Biocon
-
Dr. Reddy Laboratories
-
Fresenius Kabi
Regulatory Landscape - Table of Content
Table of contents will appear here once available.
Customer Stories

“This is really good guys. Excellent work on a tight deadline. I will continue to use you going forward and recommend you to others. Nice job”

“Thanks. It’s been a pleasure working with you, please use me as reference with any other Intel employees.”

“Thanks for sending the report it gives us a good global view of the Betaïne market.”

“Thank you, this will be very helpful for OQS.”

“We found the report very insightful! we found your research firm very helpful. I'm sending this email to secure our future business.”

“I am very pleased with how market segments have been defined in a relevant way for my purposes (such as "Portable Freezers & refrigerators" and "last-mile"). In general the report is well structured. Thanks very much for your efforts.”

“I have been reading the first document or the study, ,the Global HVAC and FP market report 2021 till 2026. Must say, good info! I have not gone in depth at all parts, but got a good indication of the data inside!”

“We got the report in time, we really thank you for your support in this process. I also thank to all of your team as they did a great job.”
